University of Wisconsin Hospitals and Clinics Madison, WI
Patrick Kittredge, MD, Adnan Said, MD, MS University of Wisconsin Hospitals and Clinics, Madison, WI
Introduction: Alpha-1-antitrypsin (AAT) deficiency is a rare autosomal codominant genetic disorder which results in liver and lung damage. AAT is a protease inhibitor produced in the liver. In ATT deficiency, the mutated AAT accumulates in hepatocytes resulting in cell injury and liver fibrosis. Lack of functional AAT leads to limited inhibition of pulmonary elastase and emphysema. AAT deficiency typically presents in early adulthood with obstructive lung disease. Two clinically relevant genotypes of AAT deficiency are PiZZ and PiMZ (PiMM is the normal state). MZ is more common and results in mild disease often in combination with other causes of liver disease. ZZ is more severe and often results in lung disease prior to liver involvement. Here we discuss a case of AAT PiZZ deficiency with delayed presentation of isolated liver disease.
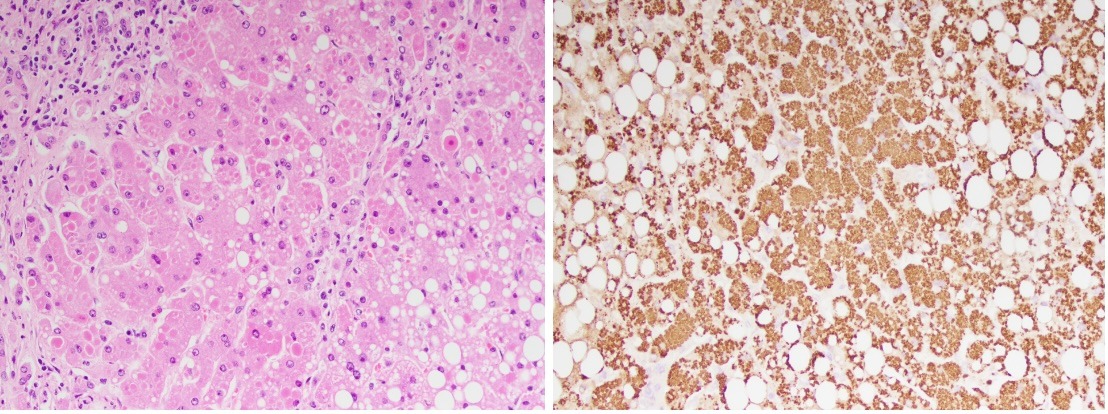
Case Description/Methods: A 61-year-old female, with an initial complaint of diarrhea, presented to a hepatology consultation with subacute progression of renal failure with newly diagnosed cirrhosis. A CT scan from one month prior showed a cirrhotic liver, ascites and portal hypertension. She had no family history of liver disease, although was diagnosed with MASLD after liver US in 2021. Initial lab workup included viral hepatitis labs, ANA, AMA, ASMA, Ceruloplasmin and AAT. Initial results revealed positive ANA and anti-smooth muscle antibody. Serum AAT level was 28 (normal 90-200). AAT phenotype resulted as PiZZ. Liver biopsy showed marked hepatocellular periportal AAT storage, consistent with the diagnosis of AAT deficiency with no evidence of MASLD or autoimmune liver disease. Renal function was deemed secondary to hepatorenal syndrome and progressed to dialysis dependence. She was able to receive a liver transplant, and is pending further evaluation for kidney transplantation.
Discussion: This patient presented in her 6th decade with ascites and renal failure secondary to hepatorenal syndrome. From symptom onset to liver transplant, 7 months had passed. Her AAT deficiency phenotype resulted as PiZZ, which is often associated with neonatal jaundice, early emphysema, and adult cirrhosis. Over 80% of adults with this phenotype develop emphysema, however this patient had no pulmonary disease on detailed testing. 20% of patients will develop cirrhosis. Given the significant heterogeneity in the presentation and natural history of ATT deficiency, it should be routinely evaluated as part of the work-up for cirrhosis independent of the presence of concomitant lung disease.
Figure: Image 1 (left): Patient’s liver biopsy specimen prior to staining Image 2 (right): Biopsy after AAT antibody immunohistochemical stain. Brown stain represents accumulated AAT within hepatocytes
Disclosures:
Patrick Kittredge indicated no relevant financial relationships.
Adnan Said indicated no relevant financial relationships.
Patrick Kittredge, MD, Adnan Said, MD, MS. P1320 - Isolated Decompensated Cirrhosis Secondary to PiZZ Alpha-1 Antitrypsin Deficiency, ACG 2024 Annual Scientific Meeting Abstracts. Philadelphia, PA: American College of Gastroenterology.