Donovan J. Kohler, MD1, Sonya Dave, MD2, Tavia Buysse, MD2, Francis Edeani, MD2 1Medical College of Wisconsin, Wauwatosa, WI; 2Medical College of Wisconsin, Milwaukee, WI
Introduction: Amyloidosis is a rare disease characterized by the abnormal deposition of insoluble beta-sheet fibrillar proteins into various tissues and organs throughout the body. The disease is most known to affect the heart, liver, spleen, and kidneys and cause a variety of associated disorders. The incidence of amyloid light chain (AL) amyloidosis is estimated at 1 case per 100,000 in western countries with 5,000-7,000 new cases diagnosed annually in the United States. Biopsy-proven light-chain amyloidosis involving the gastrointestinal (GI) tract is rare, with only 3.2% of 2,334 patients with all types of amyloidosis at one center having GI tract involvement on biopsy. In these cases, tissue diagnosis is usually obtained from duodenal or rectal biopsies.
Case Description/Methods: Our case involves a 71-year-old male with history of coronary artery disease and Parkinson’s disease who presented with an inability to tolerate solid foods due to right upper quadrant pain. He had unintentional 15-pound weight loss over four weeks, poor appetite, fatigue, and jaundice. Workup revealed rapidly worsening liver and renal function. MRCP showed heterogeneous enhancement of the hepatic parenchyma and extrahepatic biliary tree wall thickening.
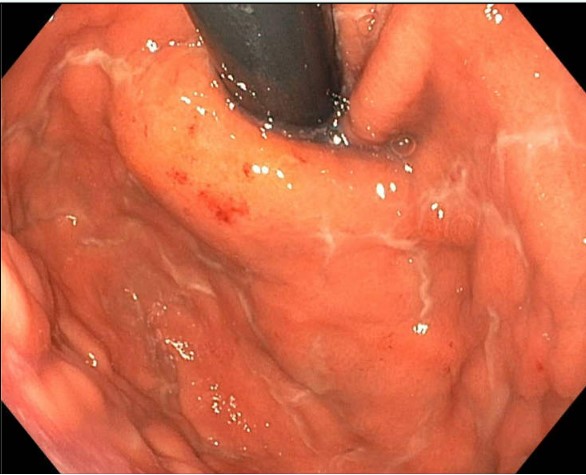
An esophagogastroduodenoscopy was performed and showed diffuse gastric and duodenal erosions and thickening of the proximal stomach and gastric body folds. Gastric biopsy histology showed chronic gastritis with fibrosis and Congo-Red stain positive for amyloidosis. Light chains were present in a 58.5 Kappa/Lambda ratio with kappa light chains of 1268. The patient was diagnosed with AL amyloidosis based on biopsy of the gastric mucosa and bone marrow biopsy confirmed plasma cell myeloma with the presence of 10-15% plasma cells by CD138 immunohistochemistry. Additionally, with the patient’s rapidly declining renal function, transthoracic echocardiogram suspicious for cardiac amyloid with classical “cherry-on-top pattern” and rising liver enzymes, it was concluded that he had renal, cardiac and liver involvement of his amyloidosis.
Discussion: Our case involved a rare diagnosis of AL amyloidosis via gastric biopsies. Endoscopic findings are non-specific and include erosions, friable mucosa, and granular appearance of the stomach and small intestine. With its poor prognosis, amyloidosis requires prompt diagnosis, especially since GI and hepatic involvement carries shorter median survival rate and elevated relative risk of death.
Figure: Figure 1. Proximal stomach with nodularity, edema, erosions, and erythema.
Disclosures:
Donovan Kohler indicated no relevant financial relationships.
Sonya Dave indicated no relevant financial relationships.
Tavia Buysse indicated no relevant financial relationships.
Francis Edeani indicated no relevant financial relationships.
Donovan J. Kohler, MD1, Sonya Dave, MD2, Tavia Buysse, MD2, Francis Edeani, MD2. P3343 - A Rare Case of Initial Diagnosis of Systemic AL Amyloidosis via Gastric Biopsies, ACG 2024 Annual Scientific Meeting Abstracts. Philadelphia, PA: American College of Gastroenterology.