University of Iowa Hospitals & Clinics iowa city, IA
Gopala Koneru, MBBS, Tomohiro Tanaka, MD, MPH, Matthew Gosse, MD, Yasameen Muzahim, MD University of Iowa Hospitals & Clinics, Iowa City, IA
Introduction: Late Onset Pompe Disease (LOPD) is a rare inherited autosomal recessive disorder characterized by deficient activity of acid alpha-glucosidase (GAA) enzyme, leading to glycogen accumulation in various tissues and subsequent myopathy. While primarily associated with progressive myopathy, LOPD can manifest with diverse clinical presentations, posing diagnostic dilemmas, particularly in adult patients. Herein, we report a case of LOPD initially masquerading as chronic hepatic injury.
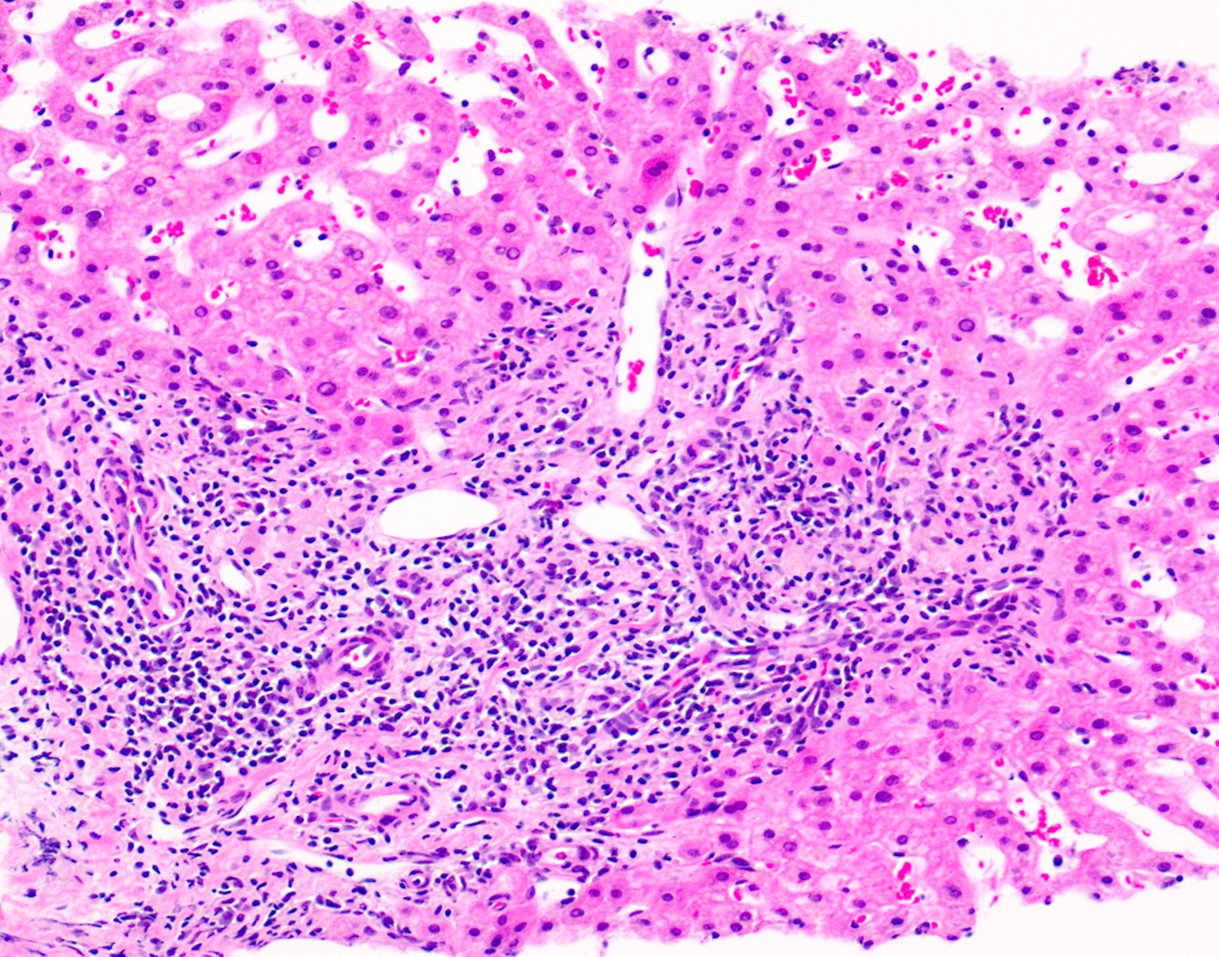
Case Description/Methods: A 32-year-old female with a complex medical history, including morbid obesity, schizoaffective disorder, IBS, and hidradenitis suppurativa, presented with elevated liver enzymes detected three years prior. Initial investigations revealed elevated Aspartate Transferase (AST) and Alanine Transaminase (ALT) levels twice the upper limit of normal, prompting further evaluation for chronic liver diseases. Work up including a liver biopsy yielded inconclusive results. The patient complained of fatigue, weight gain and persistently elevated aminotransferases and was referred to a tertiary Hepatology clinic. Further work up including initial rheumatological work was negative and subsequent repeat liver biopsy revealed a chronic hepatitic pattern of injury with minimal interface activity, mild sinusoidal dilation, and mild fibrous portal expansion (Stage 1 of 4), indicating potential progression but indeterminate for an etiology (Figure 1). In a subsequent clinic visit, she noted chronic progressive muscle aches, and she was found to have elevated creatine kinase level of 1152 U/L. Subsequent investigations uncovered nonspecific evidence of myositis and vacuolar myopathy. Genetic testing showed two pathogenic mutations in GAA which confirmed the diagnosis of LOPD, elucidating the underlying etiology of the patient's multi-organ pathology.
Discussion: The presented case illustrates the importance of considering extrahepatic causes in the differential diagnosis of persistently elevated transaminases and diagnostic challenges posed by LOPD, particularly when presenting with atypical clinical features such as chronic hepatic injury. The delayed diagnosis underscores the importance of a comprehensive and interdisciplinary approach involving Hepatology, Rheumatology, Neurology, and Genetics. Early recognition of LOPD is paramount, as timely initiation of enzyme replacement therapy with Alglucosidase alfa can modify disease progression, improve clinical outcomes, and enhance quality of life.
Figure: Figure 1: H&E-stained section shows a mild portal-based, chronic inflammatory infiltrate with focal interface activity. There are no significant plasma cells or eosinophils seen. The surrounding lobule shows mild sinusoidal dilation without significant steatosis or inflammatory activity.
Disclosures:
Gopala Koneru indicated no relevant financial relationships.
Tomohiro Tanaka indicated no relevant financial relationships.
Matthew Gosse indicated no relevant financial relationships.
Yasameen Muzahim indicated no relevant financial relationships.
Gopala Koneru, MBBS, Tomohiro Tanaka, MD, MPH, Matthew Gosse, MD, Yasameen Muzahim, MD. P4858 - Late Onset Pompe Disease Masquerading as Chronic Hepatic Injury: A Case Report, ACG 2024 Annual Scientific Meeting Abstracts. Philadelphia, PA: American College of Gastroenterology.