Irhoboudu D. Atogwe, MD1, Ahmed Alemam, MD2, Bhavna Balar, MD3 1Albert Einstein Medical Center, Philadelphia, PA; 2BronxCare Health System, New York, NY; 3BronxCare Health System, Bronx, NY
Introduction: Hemophagocytic lymphohistiocytosis (HLH) usually presents as an acute or subacute febrile illness, associated with multiple organ involvement. It primarily occurs in infants and children, though reported cases in adults are rare. This condition arises from impaired deactivation of activated macrophages and lymphocytes, leading to an exaggerated immune response. Triggers can be categorized into immune activation (e.g., viruses like EBV) or immune deficiency (such as HIV, rheumatologic diseases, and malignancy). Hepatic involvement is common, ranging from hepatitis to acute liver failure necessitating transplantation.
Case Description/Methods: A 47-year-old African American man, with a past medical history of hypertension and diabetes, presented with complaints of fevers, weakness, night sweats, loss of appetite, significant weight loss, and multiple episodes of watery diarrhea per day intermittently over 20 years, often managed with unidentified herbal remedies. He had a fever (38.7 C/101.7 F) with otherwise stable vitals. He was in no distress but his abdominal exam was notable for tenderness to palpation in the right upper quadrant. The patient exhibited no stigmata of chronic liver disease. An ultrasound of the abdomen revealed diffuse hepatic steatosis with prominent subhepatic lymph nodes. The common bile duct size was 3 mm, and the spleen was 9.7 cm. A CT abdomen/pelvis showed hepatic steatosis, and non-specific enlarged lymphadenopathy in the upper retroperitoneum and gastric hepatic ligament. Magnetic resonance cholangiopancreatography (MRCP) results were unremarkable. He received a course of broad-spectrum antibiotics, but no source of infection was identified. Epstein-Barr Virus DNA PCR was elevated to 71,171 [< 20 copies/ml], and a decision was made to proceed with a bone marrow biopsy confirming the HLH diagnosis. His final H score was 281 points, with a >99% probability of HLH. The patient was started on pulse steroids in combination with IV ganciclovir with clinical and laboratory improvement
Discussion: A high clinical suspicion needs to be maintained for the prompt diagnosis of HLH syndrome as delays can result in significant morbidity and mortality. Patients who meet more than 5 of the 9 diagnostic criteria should undergo extensive workup to identify the potential underlying etiology. They should be started on corticosteroids in addition to therapy directed toward the underlying cause, if possible.
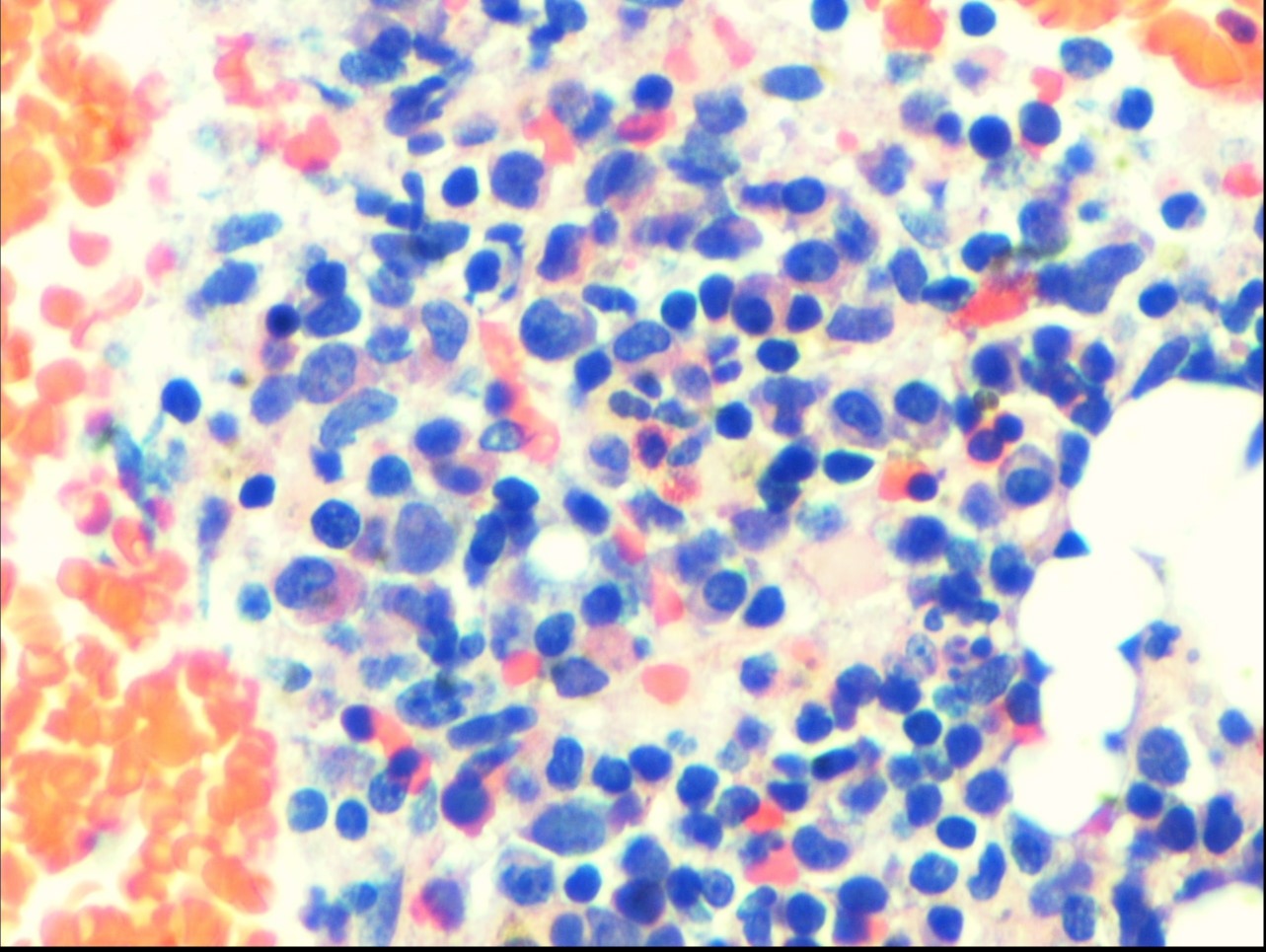
Figure: Bone marrow biopsy showing lymphoid infiltrate, frequent large histiocytic cells with overt hemophagocytosis
Note: The table for this abstract can be viewed in the ePoster Gallery section of the ACG 2024 ePoster Site or in The American Journal of Gastroenterology's abstract supplement issue, both of which will be available starting October 27, 2024.
Disclosures:
Irhoboudu Atogwe indicated no relevant financial relationships.
Ahmed Alemam indicated no relevant financial relationships.
Bhavna Balar indicated no relevant financial relationships.
Irhoboudu D. Atogwe, MD1, Ahmed Alemam, MD2, Bhavna Balar, MD3. P1383 - Better Late Than Never: A Case of Acute Hepatitis Unveils a Two Decade Undiagnosed Case of Hemophagocytic Lymphohistiocytosis, ACG 2024 Annual Scientific Meeting Abstracts. Philadelphia, PA: American College of Gastroenterology.